
用LaTeX排版化学内容核心就是两个宏包mhchem和chemfig。前者上手容易,简单易懂,适合写无机化学或是简单的有机化学,而后者功能强大却学习成本异常的高,笔者蹑足网页之间,而经过大量理解与AI的帮助,终于倔起阡陌之中(雾)。总之,本文章力求简介明晰、通俗易懂,让大家理解透彻。另外,如果有什么不懂的地方可以在评论区提出。
mhchem(无机+简单有机)
在导言区使用如下命令来引入mhchem宏包:
\usepackage[version=4]{mhchem}
基本微粒的书写
不用考虑上下标,直接将其包裹在$\ce{}$中,如$\ce{Na+}$、$\ce{Na2SO4}$等,上下标会自动标注。若该原子团中上下标连在一起,会默认全是下标,这个时候需要在上下标间隔部分用^隔开,如$\ce{SO4^2-}$。
上面三个例子编译出来的结果分别为、、。
用户只需要输入plain text就可以,而mhchem要考虑的就多了。
如果想书写只有一条单链的简单的有机结构简式,也可以用mhchem:用-=#分别表示单、双、三键,~表示氢键,键的前后不要有空格。如$\ce{CH2=CH2}$、$\ce{CH3-CH2-OH}$、$\ce{N#N}$
上面三个例子编译出来的结果分别为、、。
化学(离子)方程式的书写
规范空格的使用
为了描述方便,定义化学(离子)方程式每一个不连续在一起的东西为一个单元,如每个参与反应的物质和其化学计量数、+、气体符号、沉淀符号、方程式中间的等号……值得注意的是,化学计量数和参与反应的物质为同一个单元。每一个单元之间要用空格隔开,这样mhchem才知道这两个单元是两个不同的东西,要分开渲染,如$\ce{2H2 ^ + O2 ^}$和$\ce{2H2^+O2^}$会得到迥然不同的编译结果:、。这提醒我们虽然在大部分LaTeX语境中空格都无关紧要,但是在使用mhchem时请一定规范空格的使用!
部分特殊单元的书写
气体符号:^,等价于$\uparrow$
沉淀符号:v,等价于$\downarrow$
可逆符号:<=>T[above][below],
单向箭头:->T[above][below],
长等号:需要使用extarrows中的:\xlongequal[below]{above},
\text{中文}。demo1: 使用mhchem书写方程式
\documentclass[UTF8, 10pt, a4paper, oneside]{ctexart}
\usepackage[version=4]{mhchem}
\usepackage{extarrows}
\usepackage{amstext}
\linespread{1.5}
\pagestyle{plain}
\begin{document}
$\ce{K2CO3 + 2HClO4 = 2KClO4 v + CO2 ^ + H2O}$
$\ce{CO + CuO \xlongequal{\text{高温}} CO2 + Cu}$
$\ce{N2 + 3H2 <=> 2NH3}$
$\ce{ Li_{x}C_{y} + Li_{1-x}CoO2 <=>T[放电][充电] C_{y} + LiCoO2}$
\end{document}
编译结果:

chemfig(有机)
理论上讲,mhchem能做到的chemfig也能做到,与此同时,chemfig还有许多其他的新功能。那我们为什么还要使用mhchem呢?答案是,mhchem简明易懂、简单容易上手、自动化程度高,而chemfig代码可读性差、不容易学、编译耗时久。比如,mhchem中可以自动上下标,但在chemfig中不行,需要显式地用^、_来声明。还有诸如此类的许多问题,继续阅读你就知道了。
和mhchem一样,若非特殊声明,chemfig的所有指令都是包含在$\chemfig{}$中的。
结构式(键线式)
单链有机物
\setchemfig{debug=true}模式(debug模式)下编译的结果。我们把结构式中每一个基团叫做节点(对应键线式就是每一个起点、终点、拐点),中间的化学键被称为钩子。节点可以为任意符号、数字、字母、中文甚至为空(键线式);钩子连接了两个节点,把在代码中左边的节点称为父节点,右边的节点称为子节点。如下图中红色的方框就指示了节点:

demo2: 初识chemfig

上图的chemfig代码是:
\chemfig{CH_3-CH_2-[2]CH_3}
试着在代码中找一找,哪些是节点,哪些是钩子。[2]是钩子参数,将在后文介绍。
钩子的类别、参数
所有钩子的类别如下表:

在键线式中,其他键与双键连接时,会默认接在两条线段之间(如下左图所示)而不是我们熟悉的接在其中一条线段上,解决方案是用=^代替=,如下右图所示:(未使用debug模式编译!)
在钩子的后面紧跟一个[]即可配置其参数,顺序为[角度,键长,父节点碳序数,子节点碳序数,tikz代码]
具体解释如下:
-
角度
-
- 绝对角度
绝对角度是以父节点为参考点,钩子与x轴正方向向正方向旋转的角度值。就是如下图的

\chemfig{F-[:60]S}
像上面一样,[:绝对角度]就可以来指示键的角度。
-
- 预定义角度
每次算绝对角度太麻烦啦,你这样抱怨道。由于结构式中多为横平竖直式的,90、180、270等角度值广泛使用,chemfig也为我们预定义了许多角度值,如下图:
步长是[预定义角度]来表示键的角度。
chemfig中还有一种表示角度的方法:相对角度,要考虑的东西太多了,不建议你使用。与其“想了半天,转了八圈,还是终于转错了“,不如老老实实算绝对角度。
- 键长
相对值,推荐是0.6~0.8。在最开始设置就可以统一更改本括号内所有的键长:
\chemfig{[,0.7] CH_3-CH_2-CH_2}
\chemfig{CH_3-CH_2-CH_2}
- 父节点碳序数、子节点碳序数
若父节点/子节点有多个可以成键的原子(字母),会默认使用第一个来成键,可以在这里填入父、子的从左到右的序号,来实现诸如父节点第二个碳连接子节点第三碳之类的操作,如:
\chemfig{CH_3CH_2CHCH3-[2]CH_3CH_2CHCH3}
在未指定父子节点碳序数的情况下,编译出来的结果为:

用上述方式指定父子节点碳序数:
\chemfig{CH_3CH_2CHCH3-[2,,5,5]CH_3CH_2CHCH3}
编译出来的结果为:

- tikz代码
可以用此来自定义键的样式,这里不做展开。tikz是个无底洞,要是详细介绍的话就是另一篇文章了。
简单举个例子,在这个位置填颜色的英文名(white、blue…)就可以改变键的颜色。
全部加起来
综合以上内容,我们现在已经可以写出单链有机物了!随着代码编译,上一个子节点又会变成下一个父节点,依此类推,如下图:

所以只需要一直用A-B-C...的形式就可以写出单链有机物了,如:
\chemfig{[,0.7] CH_3-CH_2-[2]CH_2-[2]CH_2-CH_3 }

多链有机物
像写结构简式一样,在chemfig中,如果用A(-B)-C将子节点括起来,那么子节点-B就成了支链。当你在括号外再接其他节点-C时,就会以括号外的A作为父节点,如:
\chemfig{[,0.8] C-C(-[2]C)-C(-[2]C)(-[6]C)-C-C}

环
要讲环结构,我们先来看一个demo:
demo3: 苯环
苯的结构简式:
\chemfig{[,0.5]*6(-=-=-=)}
\qquad
\chemfig{[,0.5]**6(------)}

不难看出,想要几元环,就在*后放几个数字,并在后面括号内补几根键就行了。
环的编号与取代
\chemfig{[,0.5]*6(0(-0-0)-1(-1-1)-2(-2-2)-3(-3-3)-4(-4-4)-5(-5-5)-)}
如下图所示,和链一样,打一个括号就可以把支链支出去,并且通常不用考虑角度。
飞线
“主播,主播,你的钩子确实很强,但还是太吃操作了,有没有更加简单又强势、能将任意两个节点连接起来的钩子推荐一下吗?”
有的兄弟,有的。像这样能把无论有多远的节点连起来的钩子甚至所有键型都能用:
在要飞线的两个节点后紧邻着的加一个?就可以将它们连接,如:
\chemfig{[,0.8] C(-[2]C?)-C-C-C-C(-[2]C?)}

如果你想飞很多根线,可以给每一对?命一个名(仅接受a~z、A~Z、0~9):?[名字,{键型},tikz代码]
键型遵循上文提到的那张表,和其他的钩子保持一致:
demo4: 飞线大师
\chemfig{A?[a]-B(-[1]W?[a,{=},red]-X?[b])(-[7]YZ?[b,{-},{line width=2pt}])-C?[b,{~},blue]}

聚合物(链节符号)
通过自定义宏,可以在钩子上加上一个分隔符:
\chemfig{-[@{left,0.5},0.6]CH_2-[,0.7]CH_2-[@{right,0.5},0.6]}
\polymerdelim[delimiters={[]},height=5pt, depth=5pt, indice=n]{left}{right}
(由于定义了宏,需要编译两次)
解释一下:在第二行polymerdelim中定义了一个宏,其中delimiters就是你的分隔符,这里是[];height和depth是你分隔符的大小,建议先别调,编译出来后再凭感觉调;indice是聚合度,填n。第一行\chemfig中@{left,0.5}调用了这个宏,left就是左边的框框,right就是右边的框框,0.5是分隔符在钩子上的比例,填0.5就是使分隔符在钩子的中间。
如果键需要调整键角,那么请看下面的这个demo:
demo5: 复杂的聚合物
\left[\chemfig{[,0.6] W(-[:120]M)(-[4]M)(-[:-120]M)-[@{left,0.5}:-30,0.8]W(-[2]M)(-[6]M)-[@{right,0.5}:30,0.8]X-[2]Y(=[0]X)(=[4]X)-[2]X}\right]^-
\polymerdelim[delimiters={()},height=2pt, depth=5pt, indice=n]{left}{right}

只看其中的聚合物chemfig,你应该填@{left,0.5}:绝对角度。请不要尝试其他的角度表示方法,你会很崩溃的。
电子式
以前的\Lewis已经不再适用,我们需要新的charge:
\setcharge{shortcuts=true}
\charge{[circle]<Angle>:<Radius>=<EdgeItems>}{Centre}
其中
| 参数 | 含义 |
|---|---|
Angle |
边缘文本的绝对角度(单位是度) |
Radius |
边缘文本距离中心的半径,建议单位使用mm |
EdgeItems |
边缘文本的内容,可以是各种东西 |
Centre |
中心文本 |
例如:
\setcharge{shortcuts=true}
\setcharge{debug=true} %请注释掉这一行,这一行是那些花花绿绿的debug框框
\Charge{[circle]30=\:,120=$\ominus$,210=$\delta^+$}{Fe}
但是实测发现,当我们想输入化合物的电子式时,会发现这种解决方案排出来特别丑,我们就需要迂回一下,改用\chemfig中嵌套使用\charge的办法。即用\chemfig来将分子的结构确定,再在中心原子上加电子的小点点。
需要注意的是,键需要改成白色,很多间距需要自己调整。这里有一个例子:
\setcharge{shortcuts=true}
\chemfig{[,0.4]\charge{0:1mm=\:\hspace{0.2em}\:,90=\:,180=\:}{C}(-[2,,,,white]H)(-[4,,,,white]H)-[,0.5,,,white]\charge{90=\:,0=\:}{C}(-[2,,,,white]H)(-[0,,,,white]H)}
代码几乎很难看懂,对吧。编译出来的效果是这样的:
轨道表示式
直接看示例:
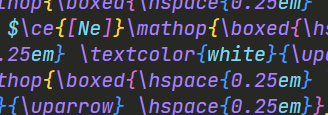
$\mathop{\boxed{\uparrow \downarrow}\boxed{\uparrow \downarrow}\boxed{\uparrow \downarrow}\boxed{\hspace{0.25em} \uparrow \hspace{0.25em}}\boxed{\hspace{0.25em} \uparrow \hspace{0.25em}}}\limits^{3d}$
其中:
| 函数 | 解释 |
|---|---|
\boxed{abc} |
将括号中的abc用方框包起来 |
\mathop |
加括号中的内容当作一个整体(一坨)来处理 |
\limits |
可以将跟在后面的^、_悬浮到前面一整坨的头顶/脚底 |
原子结构示意图
这是用tikz画的,不想讲,直接依葫芦画瓢吧:
\begin{tikzpicture}
\draw[black] (0,0) circle (4mm)node{+11};%核电荷数
\draw[red](-45:8mm) arc (-45:45:8mm)node[midway]{\colorbox{white}{2}}; %第一层电子数
\draw[red](-48:12mm) arc (-48:48:12mm)node[midway]{\colorbox{white}{8}};%第二层电子数
\draw[red](-51:16mm) arc (-51:51:16mm)node[midway]{\colorbox{white}{1}}; %第三层电子数
\end{tikzpicture}



补充:
h align=false来使聚合物的两边方框对齐以后短小的增补都会以这种形式贴出来,等积累到一定量的时候再一起改